
Termodynamik er læren om energiomdannelse i kemiske reaktioner. Man siger at enhver kemisk reaktion forløber indtil den har opnået kemisk ligevægt. Ved kemisk ligevægt sker der ikke længere en nettoreaktion, men der kan sagtens ske reaktioner i begge retninger af ligevægten – blot det sker med samme hastighed. Med termodynamik kan man forudsige retningen af en reaktion (den spontane reaktion).
Tilstandsfunktioner
I termodynamikken indgår der både tilstandsfunktioner og ikke-tilstandsfunktioner. En tilstandsfunktion er en fysisk størrelse der kun afhænger af start- og sluttilstanden – ikke nogen tilstand ind imellem. Alle tilstandsfunktioner kan skrives med et stort delta foran og på følgend måde:
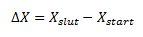
Eksempler på tilstandsfunktioner ΔH (Enthalpi), ΔG (Gibbs energi), ΔU (indre energi) og ΔS (entropi). Eksempler på ikke tilstandsfunktioner er varmen q eller arbejdet w.
En af egenskaberne ved en tilstandsfunktion er at vi med en tilstandsfunktion sagtens kunne foretage reaktionen en anden vej end normalt når bare vi ender samme sted og dette kan ske uden at værdien ændrer sig. Med ikke-tilstandsfunktioner vil værdien ændre sig når man vælger en anden reaktionsvej.
Eks.
Reaktionen fra A -> C kan både foregå direkte fra A -> C eller først fra A -> B og herefter fra B -> C. Uanset hvilken reaktionsvej vi vælger, vil de tilstandsfunktioner der findes ikke afhænge af hvilken vi vælger. Dvs. enthalpi, entropi, gibbs energi og den indre energi vil være den samme. Derimod vil varmen eller arbejdet ikke være det samme.
Et eksempel på at varme og arbejde ikke er tilstandsfunktioner ser vi når man kører en tur i bilen. Hvis vi kører en kort tur på 500 m fra A til B på 5 min. Når bilen ikke at varme motoren op. Hvis vi derimod kører en omvej stadig fra A til B men på 60 min. Dannes der meget mere varme. Det er fordi vi har valgt en anden vej.
Gibbs fri energi G
Gibbs fri energi G defineres ved:
G = H-T·S
Hvor G er Gibbs fri energi, H er enthalpien, T er temperaturen i kelvin, S er entropien. Bemærk at vi her kun har mulighed for at kende T, da man ikke kan måle direkte værdier af hverken G, H eller S.
Enthalpien afhænger af varmen som enten absorberes eller afgives under en reaktion.
Entropien afhænger af mængden af uorden i et system eller hvor mange måder energi kan fordeles på i et system.
Ved konstant temperatur får man et udtryk for ændringen i Gibbs fri energi:
ΔG=ΔH-TΔS (konstant temperatur)
Stort set alle kemiske reaktioner foregår ved konstant temperatur og derfor er denne formel meget vigtig.
Der gælder følgende for fortegenet af ΔG:
- ΔG < 0 => Reaktionen er spontan i den retning
- ΔG = 0 => Reaktionen er i ligevægt
- ΔG > 0 => Reaktionen er ikke-spontan i den retning
Systemer og omgivelser
Man definerer et system som det man betragter. Man definerer omgivelserne som alt andet (resten af universet). Der findes forskellige slags systemer:
- Åbent system: Både varme og molekyler kan passere mellem system og omgivelser
- Lukket system: Kun varme kan passere mellem system og omgivelser
- Isoleret system: Hverken varme eller molekyler kan passere mellem system og omgivelser (f.eks. en termokande hvor hverken varme eller kaffen kan passere ud).
Termodynamikens 1. lov
Termodynamikens 1. hovedsætning/lov siger at energi kan overføres mellem systemer som enten varme q eller arbejde w, men kan der kan aldrig forsvinde eller opstå ny (indre) energi.
ΔU = w+q
Altså med andre ord må ændringer i indre energi enten komme fra tilførsel af varme til et system eller tilførsel af arbejde til et system.
Forskel mellem arbejde w og varme q (work w, heat q)
Generelt er forskellen mellem arbejde w og varme q, at varme er en tilfældig form for energi (breder sig tilfældigt), som f.eks. ses når man tænder op i en kamin – her spreder varmen sig tilfældigt. Arbejde er en ikke-tilfældig form for energi, det er blandt andet det der sker når man løfter en stol. Arbejde defineres som en kraft F der påvirker et legeme i en bestemt retning s, dvs. w = F · s.
En anden forskel er at arbejde er en ordnet form for energi, mens varme er en uordnet form for energi.
Volumenarbejde / trykvolumenarbejde / pV arbejde (pressure-volumen work)
En form for arbejde er når et stempel flyttes og dermed øger trykket og formindsker volumen, dvs. en kompression. Dette er den mest almindeligste form for arbejde i et kemisk system. Når en gas komprimeres ved konstant temperatur, udøver omgivelserne et volumenarbejde på systemet og omvendt.
w = -p·ΔV
Varmekapacitet C (heat capacity)
Man skal huske på at varme og temperatur er IKKE det samme. Temperaturer en egenskab ved et system, varme er en energiform som energi kan transporteres tilfældigt med. Varmetransport sker, fordi der eksisterer en temperturforskel. Således transporteres der altid varme fra en høj temperatur til en lav temperatur. Man kan ikke måle varme direkte, men den kan beregnes udfra ændringen i temperatur, idet varmen er direkte proportionel med temperaturændringen.
q = C·ΔT
Hvor q er varmen, C er varmekapaciteten og T er temperaturen i kelvin. Enheden for varmekapacitet er J/K. Varmekapaciteten er altså den energi der skal tilføres et stof for at få temperaturen af stoffet til at stige med 1 grad. F.eks. er varmekapaciteten for metaller der leder varme let meget lav, mens den for andre stoffer der ikke let leder varme, som f.eks. gasser, er høj. Varmekapaciteten er som regel en positiv størrelse.
Specifik varmekapacitet (specific heat capacity)
Specifik varmekapacitet defineres som:
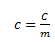
Altså er den specifikke varmekapacitet lig med varmekapaciteten divideret med massen af stoffet. Den specifikke varmekapacitet er uafhængig af mængden af stof, mens varmekapaciteten er afhængig af varmekapaciteten. Enheden for denne størrelse er J/(g·K).
Man kan kun sammenligne den specifikke varmekapacitet mellem stoffer når man skal afgøre hvilke stoffer der leder varme bedst.
Varmen afhænger altså af den specifikke varmekapacitet på følgende måde:
q = m·c·ΔT
Man kan også i symbolet for varme angive om den er under konstant volumen eller tryk ved at skrive p eller V i index: qV eller qp.
Varme kan måles i et bombekallorimeter
Enthalpi H
Enthalpien H er reaktionens varme ved konstant tryk. De fleste reaktioner sker ved konstant atmosfærisk tryk og derfor er denne størrelse meget vigtig. Under konstant tryk vil volumenet kunne ændre sig og således vil der kunne laves noget volumenarbejde i systemet. Hvis man antager at volumenarbejde er det eneste arbejde der sker vil der gælde:
ΔU = q + w = qp-p·ΔV (1)
Nu defineres enthalpi på følgende måde:
H = U + p·V
Hvilket ved konstant tryk bliver til:
ΔH = ΔU + p·ΔV (2)
Sammenholder man (1) og (2) får man:
ΔH = ΔU + p·ΔV = qp
Og således kan man altså fortolke enthalpien som værende den varme der tilføres et system ved konstant tryk.
Fortegnet for enthalpiændringen kan fortolkes på følgende måde:
- ΔH<0 => exoterm proces = der udvikles varme
- ΔH=0 => isoterm proces = der udvikles eller forbruges ikke varme
- ΔH>0 => endoterm proces = der forbruges varme
Der gælder at en enthalpiændring i én reaktionsretning skifter fortegn når man skifter reaktionsændring. Således vil en reaktion altid være exoterm i den ene retning og endoterm i den anden retning.
Standardenthalpiændringen ΔH°
Standardenthalpiændringen er enthalpiændringen der sker for en reaktion ved standardtilstand, dvs. ved koncentrationer af alle stoffer på 1 M eller et tryk af alle stoffer på 1 bar. Bemærk at en temperatur på 25oC ikke er en del af standardtilstandende.
Man er ofte mere interesseret i standardenthalpiændringen end den normale enthalpiændring, da de er nemmere at sammenligne med.
Molare standardenthalpiændring
Nogle gange opgives enthalpiændringen direkte og andre gange pr. mol stof. Begge er en form for enthalpiændring. Man kan ikke altid i symbolet for enthalpiændringen se om det er den molære eller den direkte – dette ses mest på enheden. Men man kan angive n i index for at angive at det er den molare standardenthalpiændring. Sammenhængen mellem den direkte og molare standardenthalpiændring er givet ved:
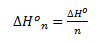
Forskellige typer af enthalpier
Man kan i index angive hvis det er en bestemt form for enthalpiændring. Der fndes mange typer:
ΔrHo reaktionsenthalpien – enthalpiændringen for en reaktion
ΔfHo dannelsesenthalpien – enthalpiændringen ved dannelse af stoffet udfra stoffets standardtilstande, dvs. stoffet er i sin mest stabile form og fysisk tilstand. Dvs. standardtilstande er f.eks. O2, C (grafit), N2 osv.
ΔcHo forbrændingsenthalpien – enthalpiændringen ved fuld forbrænding af 1 mol stof i ren oxygen gas fuldstændigt til CO2 og H2O. Er altid exoterm (negativ).
ΔbHo bindingsenthalpien – nedbrydning af en bestemt binding i et stof, så det bliver til neutrale fragmenter. Kaldes også for bindingsenergien, men et mere korrekt navn er bindingsenthalpien.
ΔlHo latticeenthalpien eller gitterenthalpien – nedbrydning af gitterstruktur for salt – kaldes også for latticeenergien eller gitterenergien.
Bemærk at standarddannelsesenthalpien for stoffer i deres standardtilstande altid er lig med 0 kJ/mol, dvs. standarddannelsesenthalpien for rene grundstoffer altid er lig med 0.
Hess’ lov
Hess’ lov siger at den totale enthalpiændring for en reaktion er konstant, uanset hvordan reaktionen gennemføres.
Med andre ord udnytter Hess’ lov at enthalpiændringen er en tilstandsfunktion og man kan derfor beregne enthalpiændringen af en reaktion hvis man kender enthalpiændringen af to andre reaktioner.
C (s) + ½ O2 (g) -> CO(g) ΔrH(A)
CO (g) + ½ O2 (g) -> CO2 (g) ΔrH(B)
C (s) + O2 (g) -> CO2 (g) ΔrH(C) = ΔrH(A)+ ΔrH(B)
Det fungerer på den måde at de støkiometriske værdier før pilen tæller negativt, dem efter pilen tæller positivt. I dette tilfælde får man altså 1 CO + 1 CO2 – 1 C – ½ O2 – 1 CO – ½ O2 = 1 CO2 – 1 C – 1 O2. Resultatet fortolker man på samme måde: De positive støkiometriske værdier skal skrives efter pilen, de negative før pilen.
Enthalpierne kan man ofte lægge sammen eller trække fra hinanden og på den måde danne C’s enthalpiændring hvis man kender enthalpiændringerne for A og B.
Bemærk at der gælder følgende formel for eks. Reaktionen A + 3B -> 2C + D.
ΔrHo =2·ΔfHo(C) + ΔfHo(D) – ΔfHo(A) – 3·ΔfHo(B) (Hess’ lov)
NB: Det er vigtigt at slå standardenthalpierne op for den korrekte fysiske tilstand (fast,flydende, gas).
Entropien S
Entropien defineres som værende det antal forskellige måder energien kan fordeles på i et system. Man kan også fortolke entropien som værende et mål for mængden af uorden. Når mængden af uorden eller mængden af måder energien kan fordeles på i system stiger, stiger entropien og omvendt.
Entropien er ligesom enthalpien en tilstandsfunktion og afhænger derfor kun af starttilstanden og sluttilstanden.
ΔSreaktion = ΔSprodukter – ΔSreaktanter
Når man øger volumen af et system, vil entropien ofte stige. Dette skyldes at molekylerne har større plads at placere sig på og tilfældigt vandre rundt på og dermed kan skabe mere uorden.
Jo højere temperatur, desto højere entropi. Dette skyldes at når temperaturen stiger, stiger hastigheden stofferne bevæger sig rundt med og dette giver mere uorden.
Der er også forskel på entropien for forskellige fysiske tilstande. Gastilstanden har den højeste entropi, flydende den næsthøjeste og faste tilstand den laveste entropi.
Termodynamikkens 2. hovedsætning/lov
Termodynamikkens 2. hovedsætning/lov siger at entropitilvæksten for et isoleret system altid vil være positiv, for at en spontan reaktion kan ske. Et isoleret system var et system hvor der hverken kunne udveksles stof eller energi med omgivelserne. Hele universet er et specialtilfælde af et isoleret system og således gælder der at entropitilvæksten for hele universet kun kan stige – ikke falde.
ΔSuniverset > 0 ΔSisoleret system > 0
Man kan vise at entropiændringen for omgivelserne er lig med den varme der tilføres omgivelserne divideret med temperaturen i kelvin.
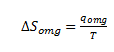
Den varme der tilføres omgivelserne er lige så stor som den varme der tilføres systemet – dog med modsat fortegn.
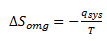
Oppe i afsnittet for enthalpi blev det vist at den varme der tilføres systemet var lige med enthalpiændringen for systemet.
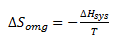
Termodynamikkens 3. hovedsætning/lov
Ved det absolutte nulpunkt er entropien af en perfekt ordnet rent krystalstof lig med 0. Dette er den 3. hovedsætning i termodynamikken.
S = 0 (Ved T = 0 K)
Det betyder at ved det absolutte nulpunkt vil mængden af uorden være minimal.
Standardentropien
Standardentropien for et stof er givet ved stoffets entropi ved 25 oC ved standardbetingelser (1 M, 1 bar). Dette kaldes for standardentropien og symboliseres ved So.
Entropiændringen
For en reaktion aA + bB -> cC + dD kan man beregne standardentropiændringen ved:
ΔSo = d·So(D)+ c·So(C)- a·So(A)- b·So(B)
Forskellige typer af entropi
Igen kalder man entropiændringen ved en reaktion hvor et stof dannes udfra dets grundstoffer for dannelsesentropien ΔfSo.
Temperatur for entropiændring
Normalt skriver man temperaturen for entropiændringen i index for at angive ved hvilken temperatur entropien er beregnet ved (Er ofte 298 K).
ΔSo298K
Standard Gibbs fri energi ændring
Hvis ΔG bestemmes ved 1 bar kaldes ændringen for standard Gibbs fri energi ændring.
Og der gælder følgende sammenhæng for reaktionen aA + bB à cC + dD
ΔGo = d·Go(D)+ c·Go(C)- a·Go(A)- b·Go(B)
NB: Ligesom det gælder for enthalpier, vil standard gibbs dannelsesenergi være lig med 0 for grundstoffer i deres standardtilstand, f.eks. O2, N2, C(grafit).
Sidst opdateret 30. maj 2023